Subscribe to Anthropic Science
Features on AI-assisted discoveries, practical workflows, and field notes across the sciences.

We’re working with world-class synthetic, computational, and analytical chemists to make Claude better at chemistry. In this post, we share our first work as part of this effort, in which Anthropic chemist, David Kamber, examines how Claude performs on a chemist’s most common analytical input, an NMR spectrum.
When working with molecules, chemists move between hand-drawn structures on a whiteboard, instrument readouts, database query strings, and the technical notations of patents and publications. Each of these representations encodes the same underlying chemistry, but each demands a different kind of fluency. A sketch of caffeine, for example, allows a chemist to spot its resemblance to adenosine, the body’s drowsiness signal, and predict that it keeps us alert by blocking the receptor. However, that same sketch cannot help a chemist tell it apart from other near-identical looking molecules.
Understanding what molecule a chemist is working with is critical. Chemistry undergirds everything from the foods and medicine we ingest to our lotions, paints, and plastics. Reroute a handful of bonds among the same atoms, and glucose becomes fructose, molecules sharing a formula but processed through entirely different metabolic pathways. Flip a molecule into its mirror image, and a sedative becomes a teratogen, as happened in the thalidomide disaster.1 Chemists’ everyday work depends on reading these signals correctly across whichever representation befits a given task.
Translating between these representations (chasing down a structure from a figure, reconciling an instrument readout against a proposed product, querying a database in the right notation) is time consuming and impossible to keep up with at scale—CAS, the largest chemistry registry, catalogs over 290 million disclosed substances and grows by roughly 15,000 new ones every day.
AI is well-positioned to take on this research burden, yet it still remains largely aspirational in the context of chemistry. Machine-learning tools have been positioned for years as transformative for retrosynthesis—the process of working backward from a target molecule to simpler precursors to plan how to build it—reaction prediction, and property estimation, but the data those tools need have been hard to come by—sparse on null-results, inconsistent in format, and locked behind paywalls at subscription journals (and in unstructured supporting information). Retrosynthesis is a case in point—capable AI tools have existed for years, but adoption is uneven, and the average academic or small-lab chemist still doesn't use them.
Even so, advancements in AI are finally reaching chemistry. Today’s frontier models are multimodal, and capable of explicit reasoning. They can read a chemical structure directly from a journal figure or hand sketch rather than depending on a pre-curated molecular database. And they can read the experimental detail of a methods section or supporting information in the form it is actually published. They can also show their reasoning step by step, which means a chemist can audit the outputs. None of this eliminates the data problem the field has been describing for years, but it changes which problems are tractable despite it.
Ultimately, our claim is a modest one: Claude is starting to meaningfully assist chemists with the daily translation, recall, and integration work that complements their judgment, and we plan to keep extending its helpfulness. Today we are publishing the first white paper in the effort to accelerate this work. It tackles a chemist's most common analytical input: an NMR spectrum.
Full version can be found here
Nearly every small molecule—drug, pesticide, dye, fragrance, polymer, DNA or protein subunit, and functional inorganic or solid-state material—exists because a chemist determined its structure. Given that these molecules cannot be seen with microscopes, chemists must rely on spectral analysis, probing a molecule with light, radio waves, or magnetic fields. The way a given molecule absorbs, emits, or deflects this energy gives chemists a pattern, or spectrum, with which they can elucidate its structure.
NMR spectroscopy—one of the canonical techniques chemists rely on for this—is one of the most time-consuming steps in synthetic chemistry; for every compound, a chemist has to match each peak in the spectrum to an atom in the proposed structure by hand. For this white paper, we tested how Claude fared against the dedicated NMR software chemists rely on today. We measured three Claude models (Opus 4.7, Opus 4.6, Sonnet 4.6) against ChemDraw and MestReNova on 20 compounds drawn from synthetic chemistry preprints published after the models’ training cutoff so as to avoid selection bias. Both ChemDraw and MestReNova do forward prediction, using a drawn structure to simulate what NMR spectrum will be produced. In addition to forward prediction, we also wanted to see whether Claude could go the other direction—starting from an experimental spectrum and proposing the structure behind it. This is the harder task, and the one existing software currently leaves to the chemist.
To set up our assessment, we pulled 20 compounds from ChemRxiv preprints2 posted after the models’ training cutoff, taking the first fully characterized novel molecules from each paper. The 20 span four structural families, five compounds each, with each family selected because it involves a different category of NMR challenge. Each tool was given the structure encoded as a SMILES string—the line-of-text notation chemists use to input a molecule to software—and was asked to predict where every hydrogen and carbon peak would fall along a 1D NMR spectrum (a horizontal axis measuring chemical shifts in ppm, parts per million). Given that NMR samples are dissolved in a liquid, and that the choice of solvent (chloroform, DMSO, etc.) moves the peak positions slightly, each tool was told to predict the spectrum in whatever solvent the chemists had used in the published paper.
Because a language model’s output varies between runs, each Claude model was queried three times per compound and averaged; ChemDraw and MestReNova return the same answer every time and were run once. We then paired each predicted peak with its experimental counterpart and measured the gap in ppm. These landed within the window a chemist would call correct—±0.20 ppm for hydrogen or ±1.0 ppm for carbon.
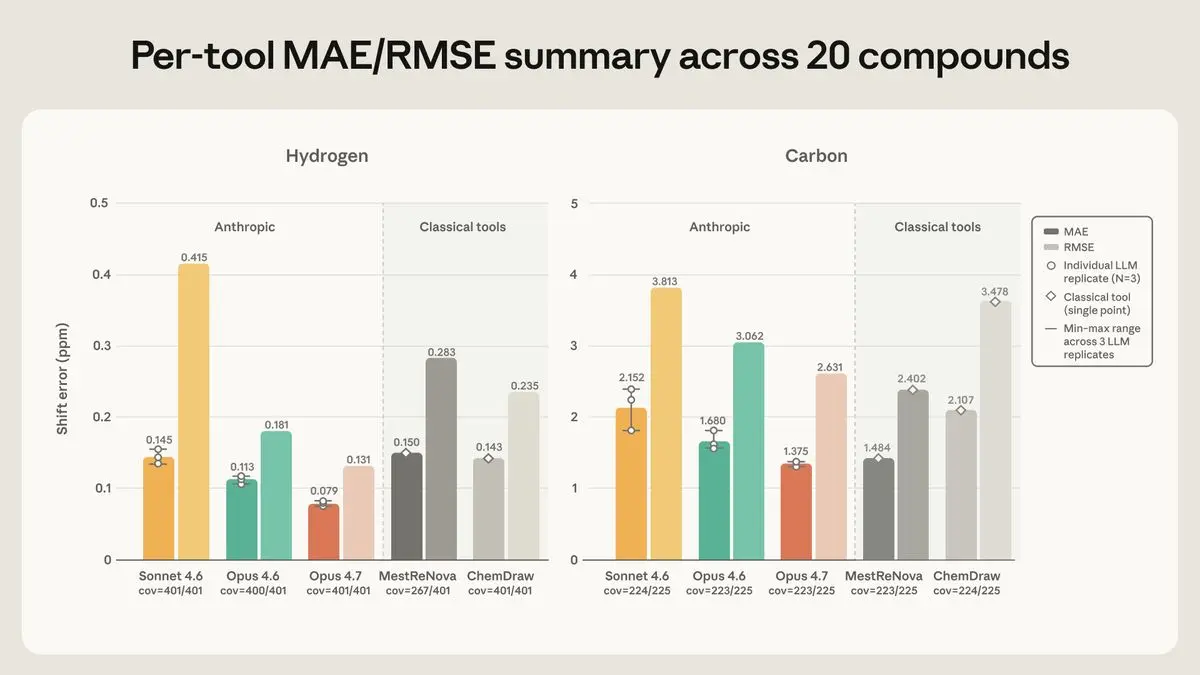
On hydrogen, Opus 4.7 was most accurate, with an average error of ±0.079 ppm—well under half the tolerance window—and the highest share of peaks landing inside it. On carbon, Opus 4.7 and MestReNova were effectively tied, at ±1.37 and ±1.48 ppm; the remaining tools kept the same rank order on both elements. Opus 4.6 was predictably middling, and Sonnet 4.6 was the weakest. The gap between them was most evident on a single notoriously difficult hydrogen—an NH proton in the chloropyridazine family whose true position falls in a narrow band between 6.8 and 7.9 ppm. Opus 4.7 placed it slightly low but consistently so; Opus 4.6 scattered its guesses across several ppm; Sonnet 4.6 put it in the 10–13 range, well outside where it actually appears.
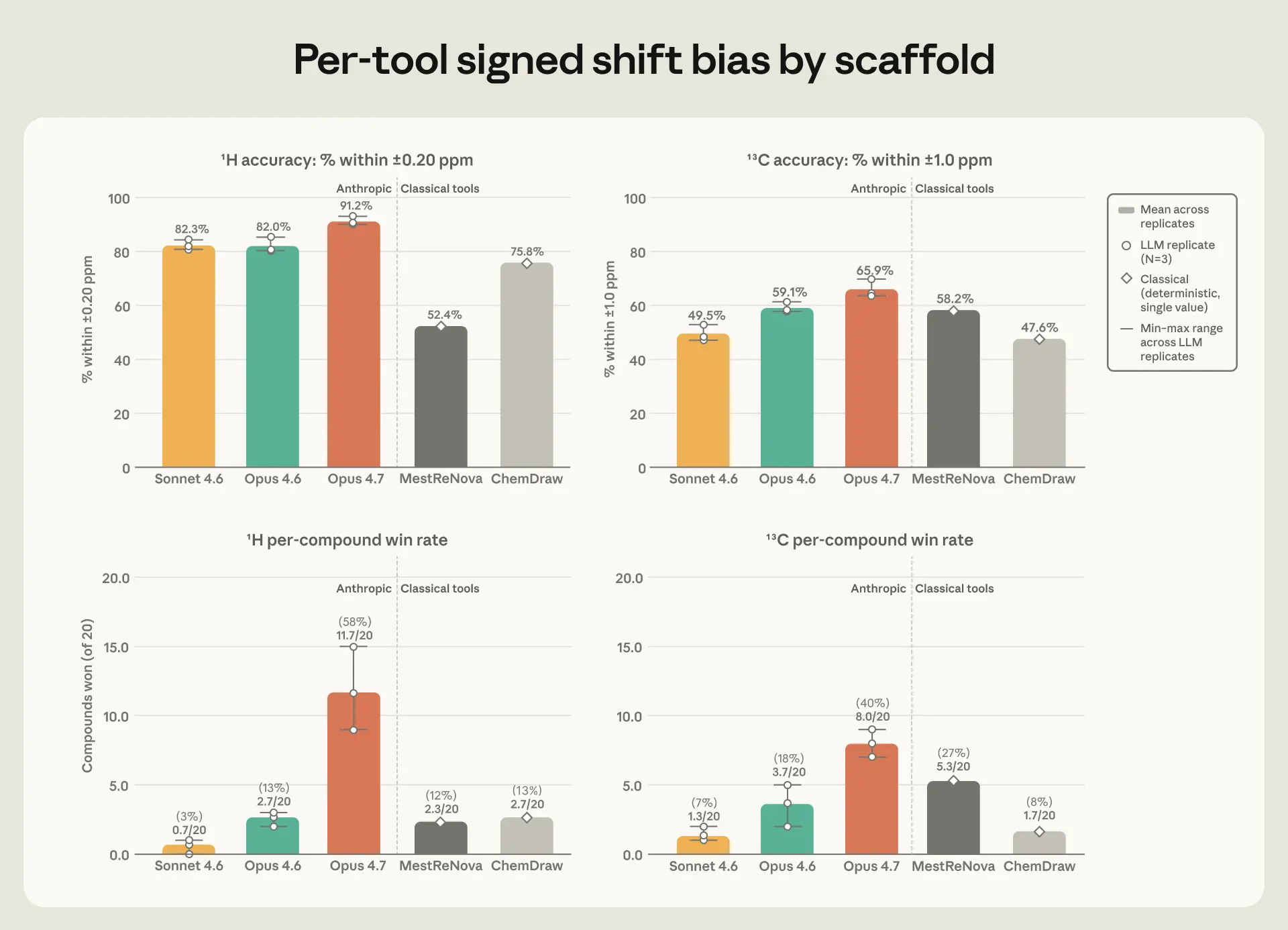
While Opus 4.7 performed fairly comparably to ChemDraw and MestReNova, the gap was wider on predicting the shape taken by a hydrogen’s NMR peak and how far apart the peaks sit, features which also contain structural information a chemist reads alongside position. Opus 4.7 matched the experimentally reported splitting pattern more often than any other tool, and all three Claude models predicted the sub-peak spacing to within half a hertz roughly 80% of the time—against 26 to 35% for ChemDraw and MestReNova. Opus 4.7 was also the most consistent across its three repeat runs: its average error varied less from run to run than the margin separating it from the next-best tool.
From there, we evaluated inverse prediction (structure elucidation): could we determine the structure of a molecule from its spectrum? We gave Opus 4.7 15 elucidation problems and asked it, three times each, to propose up to three ranked candidate structures. Each supplied the compound’s exact molecular formula (from high-resolution mass spectrometry) and its hydrogen and carbon NMR spectra. The fifteen were split by difficulty. The eight simpler targets—single-ring or two-fragment molecules—were posed with only the formula and spectra. The seven denser targets—fused rings, spirocycles, and similar—were accompanied by one additional hint: the structure of the starting material that had gone into the reaction.
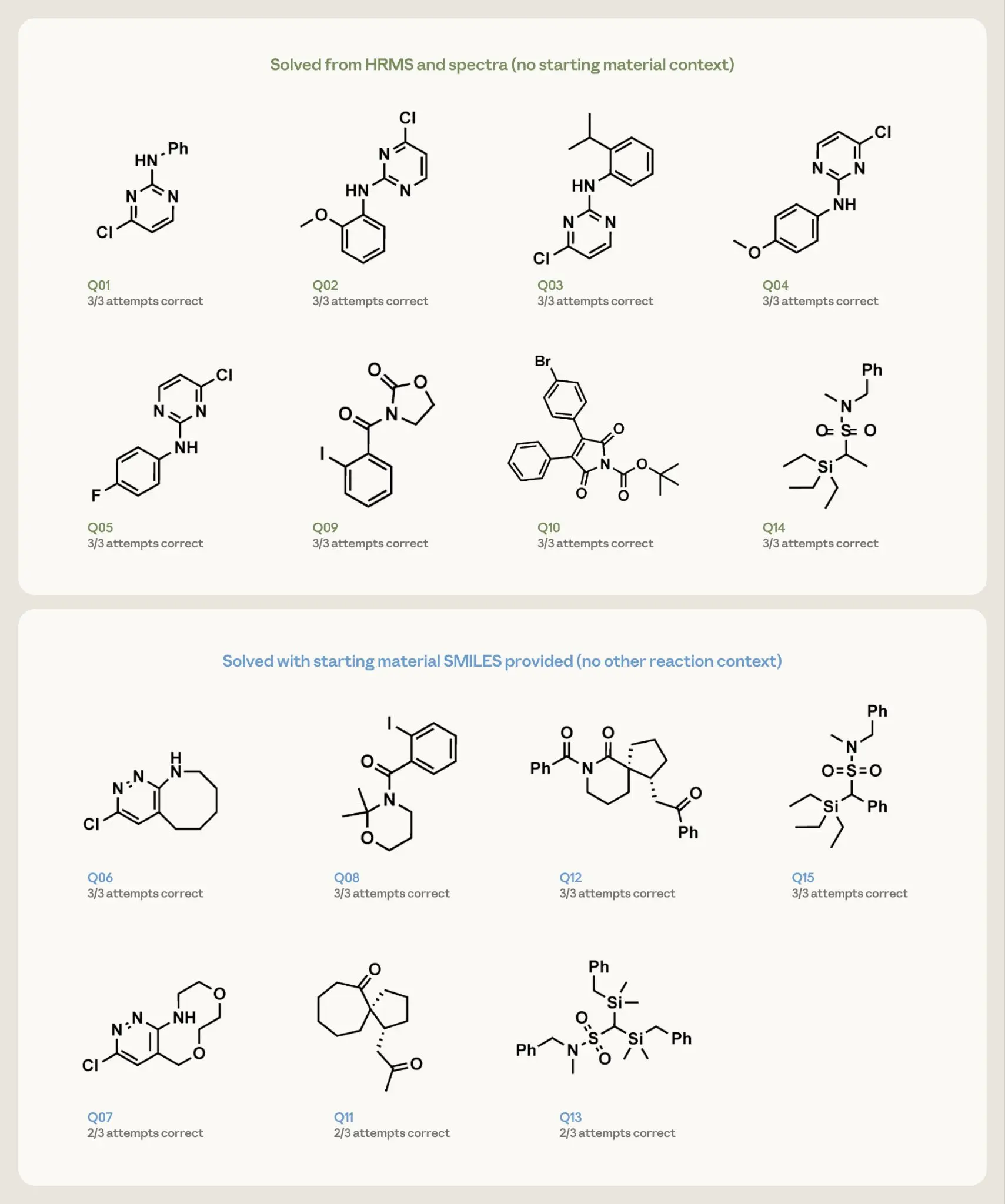
Opus 4.7 recovered all eight simpler structures on every attempt from spectra and formula alone. On the seven harder targets, given the starting-material hint, it returned the correct structure on all three runs for four of them and on two of three runs for those that remained.
Ultimately, we found that for routine data prediction Opus 4.7—a general-purpose model without chemistry-specific fine-tuning—is now as good as or better than ChemDraw and MestReNova on average. Additionally, Claude can also work the problem in reverse, proposing a structure from NMR data alone. Dedicated structure-elucidation software has existed for decades, but it typically requires 2D NMR (a spectrum with two axes, and the output is a contour map rather than a row of peaks), specialized training, and licensed tools. Claude does it from the same high-resolution mass spectrum and 1D peak list a chemist would paste into a chat, with no setup.
This assessment shows us that a general-purpose model can be competitive with NMR software and even make 1D inverse elucidation tractable. But there are a handful of noteworthy limitations.
Ideally, we would see how these numbers hold up across several hundred compounds spanning 20–30 scaffold classes, with at least 15 compounds per class so that within-class variance can be separated from between-tool differences. We would also evaluate NH-active heteroaromatics beyond chloropyridazines, assess the untested solvents, and conduct versions of both tasks that draw on 2D experiments.
As we continue to improve Claude’s performance in chemistry, we are focusing specifically on a handful of bottlenecks that slow chemists down the most.
These are not all on the same maturity curve. Where spectral analysis is far enough along to benchmark, others, like retrosynthesis planning, are still being scoped. As we get a better understanding of these bottlenecks, we will share where current models excel, and where they still fall short. Our ultimate goal is to ensure that working chemists know where Claude can save them time and where they still need to rely on their own expertise.
We are expanding the AI for Science program to more explicitly support chemistry research. If you are a researcher working on a problem where Claude could plausibly help, especially one that involves the kinds of multimodal reasoning we have described, we would like to hear from you at scienceblog@anthropic.com, or through the AI for Science application.
In project Fetch, we examined how humans can use models to get robots to perform complex tasks. Now, we investigate many models on a large variety of different robotics tasks in simulation, to see how good models are at controlling robots themselves.
Read moreFeatures on AI-assisted discoveries, practical workflows, and field notes across the sciences.