Subscribe to Anthropic Science
Features on AI-assisted discoveries, practical workflows, and field notes across the sciences.

Written by Laura Luebbert. Based on research by Ferdous Nasri, Sarah Gurev, Patrick Varilly, Krithik Ramesh, Nuala A. O’Leary, Jonah Cool, Bernhard Y. Renard, Pardis Sabeti, and Laura Luebbert.
In this post, Laura Luebbert argues that we need to make biological data infrastructure more agent-friendly. As a case study, she and her team tasked scientific research agents (Claude, Biomni Open Source (Biomni OSS)1, Edison Analysis,2 GPT) to retrieve the sequence data from NCBI Virus, a database virologists use for tasks such as surveillance and diagnostic assay development. Even the strongest models did not consistently achieve the level of accuracy required for reliable dataset construction. But accuracy rose to nearly 100% once she and her team added gget virus, a deterministic retrieval layer. The broader lesson for scientific agents is that deterministic retrieval tools are (currently) crucial to making agent workflows more reliable, and biological databases will need to be designed with agents in mind as scaled users.
Using AI agents to navigate biological data infrastructure is like driving through an old city that was designed before cars: the infrastructure may be beautiful and even thoughtful, but it’s full of narrow, winding streets that are difficult for modern vehicles to navigate (idiosyncratic file formats, scattered databases, and one-off retrieval scripts).3 You can retrofit the city with traffic signs, parking lots, and the occasional widened road, but the basic layout remains hard to navigate because it was designed for a different mode of conveyance. Software infrastructure, by contrast, was basically made for the needs of cars (agents): paved roads, clear lanes, standardized signals, and systems designed for fast travel from start to finish (version control, well-documented APIs, and package managers).
As a result, coding agents have advanced much more quickly than biological agents. Software commonly provides structured digital workflows and reliable interfaces, whereas the computational biology infrastructure needed for data retrieval and validation is often brittle, heterogeneous, and process-dependent. The tools with which we navigate them are necessarily bespoke and tuned to defined domains or hypotheses. Moreover, software provides testable outputs that can be quickly compiled and validated (e.g., resolving a GitHub issue by generating a patch that passes the project’s tests), whereas biology offers few simple and verifiable yet meaningful rewards.
Thus, the bottleneck for biological agents is not only reasoning but the absence of widespread deterministic execution layers for querying biological data. A scientist can express their intent (e.g., find all human kinases with this domain and pull their structures), but agents often lack a dependable way to access the databases containing the information they need.
In biological and scientific workflows, even small errors can have severe consequences. Retrieving coordinates from the wrong genome build, for example, can invalidate the downstream biological interpretation. So can mixing RefSeq and GenBank records without intending to, treating partial genomes as complete genomes, confusing segment names in segmented viruses, or missing relevant records because of inconsistent metadata fields. The beauty and challenge of research is that the details are often of critical importance.
Like driving through an Italian hill town, it does not matter how powerful the car is if the streets are too narrow, the turns too sharp, and the route depends on local knowledge. If we want agents to help with scientific discovery, from outbreak response to drug design to biological modeling, we need to build biological data infrastructure that they can navigate as reliably as humans do.
This mismatch between agent needs and human-built tools is not unique to biology. The same friction emerges wherever agents are inserted into environments designed solely for human use.
A few months ago, Andrej Karpathy gave a talk about software in the era of AI and ended up griping about something that sounded all too familiar. He had vibe-coded a small web app, but when he tried to make it real (authentication, payments, deployment), he lost a week clicking around in browser dashboards.
As he summarized, “The code was the easiest part! Most of the work was in the browser, clicking things.” Documentation kept telling him to “go to this URL, click on this dropdown.” His conclusion was that nobody should have to do this. Instead, we must build for agents.
Karpathy had experienced something new within the world of software agents that biology researchers have been struggling with for a long time: the pain of trying to make intelligent systems operate in environments built around heterogeneous information, implicit conventions, and humans clicking through browsers.
Long before AI agents, computational biologists and geneticists had already begun to produce tools for traditional computational biology, which chipped away at this problem. Packages like Biopython, BioPerl, BioJulia, Entrez Direct, BioMart, gget, and many other workflow libraries are all efforts to move biological data out of browser interfaces and into places where researchers can compute on it directly.
The problem is that biological data does not live in a single database with a single interface. It is a messy network of roads, each with its own identifiers, conventions, formats, filtering logic, and degree of programmatic access. Some data are straightforward to access programmatically. Others, not so much.
Virology, in particular, is one of the harder cases. Research workflows from vaccine and diagnostic assay design to building training data for protein models often begin by retrieving sequences from NCBI Virus, a collection of viral sequence records from GenBank, RefSeq, and the international INSDC ecosystem, including Pathoplexus, behind a searchable web interface. As researchers building tools for viral outbreak surveillance, we know firsthand how much expert knowledge is hidden behind these retrievals. In virology labs, dataset curation instructions for NCBI Virus are often passed around as long lists of complex filters that users must manually reproduce in the web interface: exactly the kind of browser-clicking workflow Karpathy was complaining about.
The current outbreak of Ebola disease caused by Bundibugyo virus in the Democratic Republic of Congo is a stark example of why streamlined viral data access can have real-world, life-or-death consequences. On May 14, 2026, INRB Kinshasa in the DRC analyzed 13 blood samples and confirmed Bundibugyo virus disease in eight the next day,4 after which an Ebola outbreak was declared. By May 29, the WHO had reported more than 1,000 confirmed and suspected cases in the DRC, including more than 200 deaths. Researchers also generated the first near-complete outbreak genomes, helping establish that the outbreak was caused by a new spillover event.
These genomes present public health officials with three urgent questions. First, how different is this outbreak virus from Ebola viruses seen before? Second, can existing diagnostics still detect it? And, third, will existing therapeutics still protect against it? Answering these questions requires comparing the new genomes against historical Ebola genomes available through NCBI Virus and Pathoplexus (which synchronizes into NCBI Virus). But rather than this being easily automatable, the first steps in this analysis involve manually clicking through a web interface, reproducing complex filters by hand, and hoping the resulting dataset is complete and correct.
The reason this workflow is so hard to automate is that much of NCBI Virus’s filtering logic lives only in this web interface. This is annoying for humans and terrible for agents. If a researcher wants every SARS-CoV-2 sequence released in 2025 that contains the surface glycoprotein, that may take a seasoned virologist a few clicks in the browser. But programmatically, it can require a multi-hundred-line script gluing together multiple APIs (REST, Datasets, E-utilities), retrieving results page by page, reconciling identifiers, and downloading hundreds of gigabytes of data, only to throw most of it away after local filtering.
Even if a resource has an API, it can still be difficult for agents to use reliably for a variety of reasons, for example if the API does not expose the same filtering semantics as the web interface, if metadata fields are poorly documented or inconsistently standardized, if identifiers change across sources, or if “the right answer” depends on conventions that expert humans know but machines have to infer.
To better understand the challenge of bridging agents to databases, we developed a test for how capable state-of-the-art scientific research agents (Claude, Biomni OSS, Edison Analysis, GPT) are when asked to retrieve viral sequences from NCBI Virus using the infrastructure available today. Our benchmark, VirBench, includes 120 realistic viral sequence queries spanning 40 pathogens with manually verified ground-truth counts. The queries reflect tasks that appear in viral surveillance, diagnostic assay design, and protein model training-data construction. For example, one query asked agents to “retrieve viral sequences from NCBI for TaxID 3052462 (Orthoebolavirus zairense (ZEBOV)) that adhere to the following criteria: host organism: human, geographic location of sample collection: Africa, collected on or after 01/01/2014, collected on or before 06/20/2014, minimum sequence length: 15,200 bases, maximum 1,900 ambiguous characters (N’s), exclude lab-passaged samples.”
When agents were left to solve these queries on their own, performance varied widely across systems and improved substantially in newer frontier models. However, even the strongest models did not consistently achieve the level of accuracy and reproducibility required for reliable dataset construction. Claude Sonnet 4, Claude Opus 4.7, Biomni OSS, Edison Analysis, GPT-5.2-pro, and GPT-5.55 achieved mean accuracies ranging from 16.9% to 91.3%. For these data retrieval tasks, the bar is effectively 100%: in some cases, a missing or incorrect record could determine whether a diagnostic assay seems to cover circulating diversity, or whether an outbreak is inferred to have started weeks earlier or later than it did. In addition, the same model often produced substantially different answers when asked the same question three times, undermining both the accuracy and reproducibility required for reliable scientific workflows. For the example Ebolavirus query above, Sonnet 46 returned 106 sequences in one run (expected: 266), 15 in a second run, and 5 in a third, despite receiving an identical prompt each time.
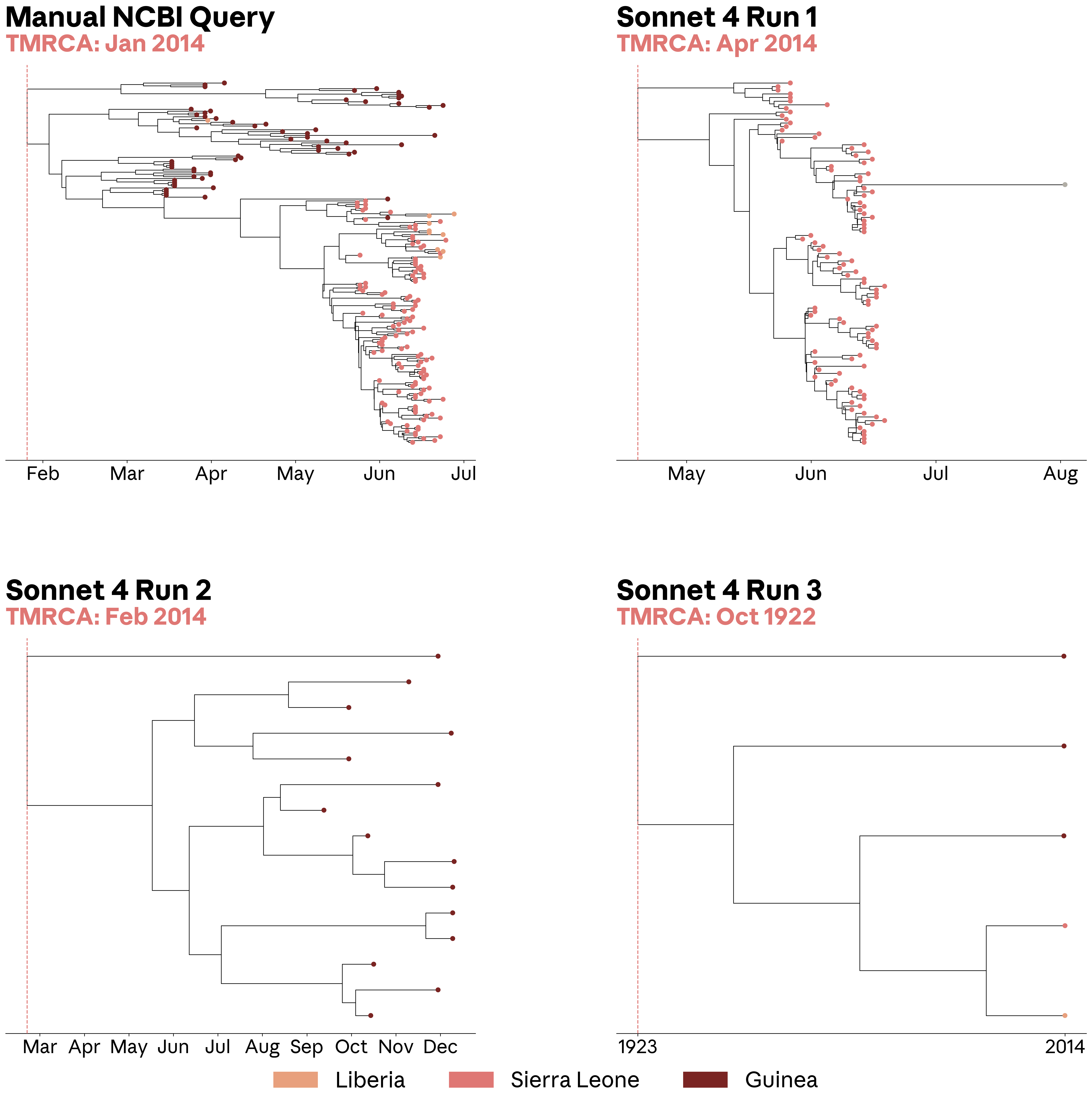
Inconsistencies like this have consequences for downstream analyses. We used the query shown above to retrieve Ebolavirus sequences and build a phylogenetic tree, a standard analysis for reconstructing how viral samples are related during an outbreak. One important quantity we can get from phylogenetic trees is the estimated time to the most recent common ancestor (TMRCA). This is the inferred root date of an outbreak, which can alter conclusions about when and where a virus originated, as well as how long a virus was circulating. In this case, a tree built from a manually curated NCBI Virus sequence set recovered a January 2014 TMRCA, consistent with previous reports (95% highest posterior density spans 27 January to 14 March) for the 2014 Ebolavirus outbreak. By contrast, two of the three sequence sets retrieved by Sonnet 4 were visibly incomplete, including one tree that pushed the inferred TMRCA back to 1922. The remaining dataset (run 1) looked superficially plausible, but failed to retrieve sequences from Guinea and shifted the estimated TMRCA to April 2014, changing the inferred timing of the outbreak.
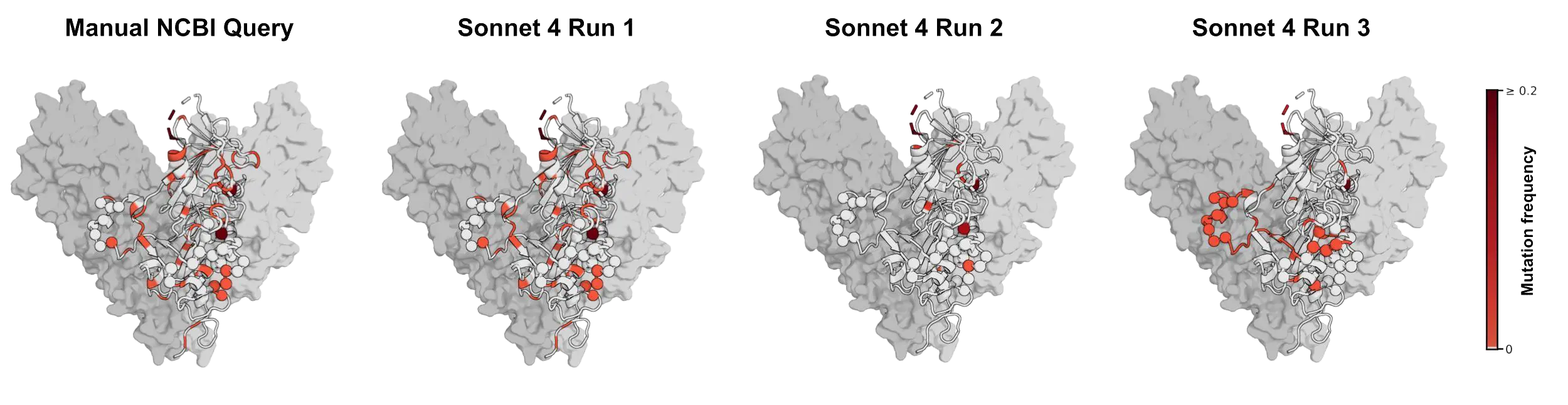
The variability between NCBI Virus retrieval attempts can also affect conclusions about therapeutics. We retrieved Ebolavirus glycoprotein sequences to examine the epitopes bound by maftivimab and MBP134, antibody therapeutics developed against Zaire ebolavirus and WHO priority treatment candidates in the ongoing Ebolavirus outbreak. We asked whether mutations have previously arisen in the regions these antibodies target across related Zaire ebolavirus sequences. This kind of analysis can give researchers a sense of whether a treatment will continue to protect patients as the virus evolves. If the underlying sequences are incomplete or incorrectly fetched, it can throw off their conclusion. In our example, sequences retrieved by Sonnet 4 came close to the results obtained through a manual NCBI query on its first attempt. On a repeat run, it missed most of the mutated residues. And, on the third run, it highlighted a different set of residues, producing three different impressions of the variability in these target regions.7
Both of these examples illustrate a broader pattern in science: details that look like minor retrieval choices can change the biological conclusion. In this case, the inconsistent model performance in viral sequence retrieval and the nature of the failure modes highlighted that most variation was attributable to infrastructure shortcomings. Agents under-counted when they failed to retrieve large result sets, and over-counted when filters were applied incorrectly. For example, the largest deviations from the expected counts occurred for viruses with large numbers of available records, including Influenza A, HIV-1, and SARS-CoV-2, where stopping partway through retrieval and incorrect downstream filtering can substantially distort the final dataset. They also struggled with metadata fields whose meaning depends on context, convention, or where information happens to be stored. Performance degraded as queries became more complex, especially beyond three or four simultaneous filters.
Ultimately, the agents often understood the task well enough to attempt it, but they lacked a machine-actionable way to carry it out, verify it, and repeat it. The resulting answers could look plausible while still being wrong, which is especially dangerous because sequence retrieval is usually the first step in a much longer biological workflow.
For a more thorough explanation of VirBench and gget virus, read the preprint.
To turn viral data retrieval into something that agents and humans could call directly, we developed gget virus in collaboration with researchers at NCBI. At first, this seemed like it might be a simple matter of connecting to the right API calls. In practice, it was much harder: NCBI Virus is a portal over multiple underlying resources, including internationally synchronized sequence databases maintained across the United States, Europe, and Japan, so answering a seemingly simple query often requires piecing together information from several places.
To reproduce the behavior of the NCBI Virus web interface, gget virus has to coordinate across the different systems underneath it, including the REST, Datasets, and E-utilities APIs. gget virus decides which filters can be applied through these existing APIs and which have to be checked locally because the web interface exposes filtering behavior that is not available from a single programmatic endpoint. It handles batching so that large result sets, such as for SARS-CoV-2 and Influenza A datasets, are retrieved comprehensively rather than arbitrarily cut off. When filtering depends on additional information stored in a separate database, such as GenBank records that indicate whether a sequence contains a particular viral protein, gget virus retrieves those records, uses them to apply the filters, and preserves the relevant GenBank information in the final output. It then returns standardized outputs that are readable by both people and machines, with detailed logs that show how the final result was produced.8
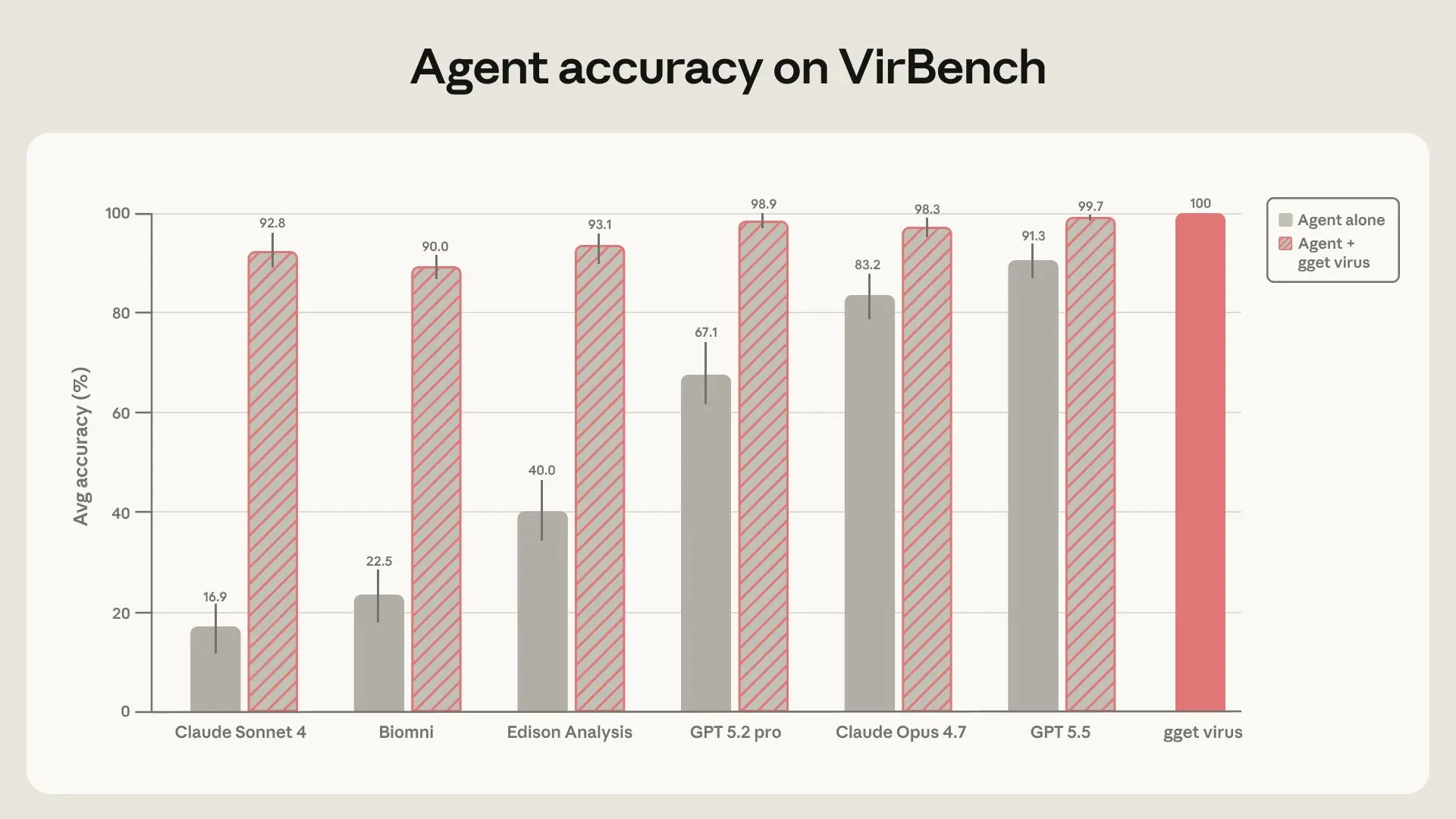
When we gave the agents access to gget virus, accuracy rose above 90% for all agents, peaking at 99.7% for GPT-5.5. Run-to-run variability was largely eliminated, and the performance gap between models narrowed dramatically. In other words, adding a deterministic retrieval layer made model choice much less important. This is especially consequential given that reliable dataset construction should not depend on access to the newest or most expensive model, or on knowing which model works best for a given database. Instead, cheaper models paired with the right tool reduce variability and enable wider access.
gget virus makes existing agents more reliable for viral data retrieval by translating a complex, browser-based retrieval workflow into an accurate and reproducible interface. Returning to our walkable city analogy, it’s like we added a highway tunnel underneath the pedestrian infrastructure, complete with on- and off-ramps, smooth-moving interchanges, and exit numbers tied to known mile markers.
We want models to be creative when they generate hypotheses, design experiments, or reason about mechanisms. But the layer underneath that creativity—gene identifiers, schemas, retrieval logic, coordinate systems, metadata conventions, and data access paths—has to be boringly reliable (or in other words, deterministic). gget virus is one example within a broader set of efforts to build these context engines: reliable, agent-accessible infrastructure for biological data. Other efforts are emerging from AI-for-science systems, many of which rely on model harnesses that connect agents to biological data sources, including ToolUniverse, Edison Scientific’s Robin, Biomni, and related biomedical agents. The challenge is figuring out where that determinism belongs and how to build it.
Work on connectors and harnesses becomes more fraught when we consider how quickly model capabilities are changing. If we draw the model curve forward from the results above, it’s easy to imagine a (very near) future in which the benefit of tools like gget virus approaches zero: agents become good enough to navigate messy portals, reconcile identifiers, paginate correctly, and recover from failures on their own. In that world, harnesses may not be needed. Still, even if an agent can do it, that doesn't mean the task should be handled (and reinvented) by an agent every time. A model that can fight its way through a confusing bioinformatics workflow may still be too expensive, too slow, too hard to audit, or too difficult to trust for routine scientific work. And if agents do eventually make today’s harnesses obsolete, the lesson for biological databases holds: we need to keep agents in mind as we think about our users, and we need to build for scale.
We thank Xander Balwit, Ethan Dyer, Stuart Ritchie, Rebecca Hiscott, Alyssa Morrow, Keir Bradwell, Eric Kauderer-Abrams, Jonah Cool, Andrej Karpathy, Patrick Varilly, Cesar Arze, Blake Lash, Philine Guckelberger, Nisha Gopal, Elliot Hershberg, Pardis Sabeti, and Jonathan Feldman for their thoughtful feedback, careful edits, and helpful conversations that improved this essay.
We are especially grateful to Sarah Gurev and Gage Moreno for their help in developing and performing the example virology analyses, and to Ferdous Nasri and Krithik Ramesh, who made substantial contributions to the ideas, framing, and writing of this essay.
In project Fetch, we examined how humans can use models to get robots to perform complex tasks. Now, we investigate many models on a large variety of different robotics tasks in simulation, to see how good models are at controlling robots themselves.
Read moreFeatures on AI-assisted discoveries, practical workflows, and field notes across the sciences.